Initial Draft – Under Construction – The parameterisation and implementation of dihedral angles are yet to be included.
This beginner’s tutorial will demonstrate how to modify a wildtype protein with post-translational modifications (PTM). I will be using the crystal structure of wildtype (i.e. unmodified) collagen from an earlier publication where I went through the exact same process. Per the linked publication, I’ll add an advanced glycation end-product crosslink between two of the collagen polypeptide chains. It’s possible to replicate many of these steps with any protein and any post-translation modification. What I won’t de discussing
Prerequisites
You’ll need at least the following:
- GROMACS simulation software suite.
- Avogadro for modifying structures.
- A protein starting structure geometry (and topology file will be helpful .itp) in .gro/.pdb (pdb2gmx helps shift from one to the other, however, it will not recognise the PTM structure unless it’s been added into the corresponding forcefield – more on that later).
- Details on the PTM structure, topology, forces and charges:
- The structure on paper or a cartesian coordinate list. Having derived this using quantum chemistry software, the energy minimised structure becomes the PTM equilibrium structure. We plug these into our GROMACS forcefield files and we do our best to insert this structure into our starting structure geometry (.gro) file using Avogadro (or something similar).
- Bonded forces constants for angles and lengths appropriate for the overall force field. Dihedral bond information is hard to come by and doesn’t collapse out of the Hessian of a QM calculation. I would recommend seeing what the existing force field comes with but this will depend on how unusual your PTM structure is compared with known topologies within the forcefield.
- Atom charge information. I’ll be modifying the Amber force field family so my partial charges were derived using the R.E.D Server.
I will be using the structure, force constants and atom partial charge values from the QM optimised structure of the oddly named DOGDIC crosslink from my preprint.
Preparing the forcefield
You can’t get away from modifying at least some small part of the forcefield files. We’ll be adding in a new residue name to the geometry and topology file and grompp will always look for this in the corresponding topology data files. It’s always worth putting in the extra mile and preparing a complete set of forcefield files. That way pdb2gmx can shift any modified .pdb file into a GROMACS file whilst generating the appropriate .top (itp) file. Preparing the topology will involve a lot of work in the ./share/gromacs/top/amber99sb-ildn.ff/ directory.
Preparing the forcefield files
The QM optimised structure contains the bond lengths and angles at equilibrium. However, fitting that particular structure into the whole geometry may not be possible so we’ll rely on a series of energy optimisation steps and MD trajectories to account for the changes we’ll make during construction. Sadly, unless you’ve derived your force constants with software such as ForceGen, you’ll need to measure the bond distance between every atom pain and the angle between atom triplets.
Firstly, build your structure on paper or a chemical software package. This is the structure I’ll be attaching between two collagen polypeptides. You’ll notice several features. Each atom is uniquely labelled and are used as atom types in the atomtype.atp file. I have marked, in red, the special bond used to link two residues by some part that is not the peptide backbone. In this example, DN2 and DC13 form the bond. I’ve marked in blue the bond used to attach residues along a backbone chain, and between those bonds and the special bond we have two residues: DOG1 and DOG2.
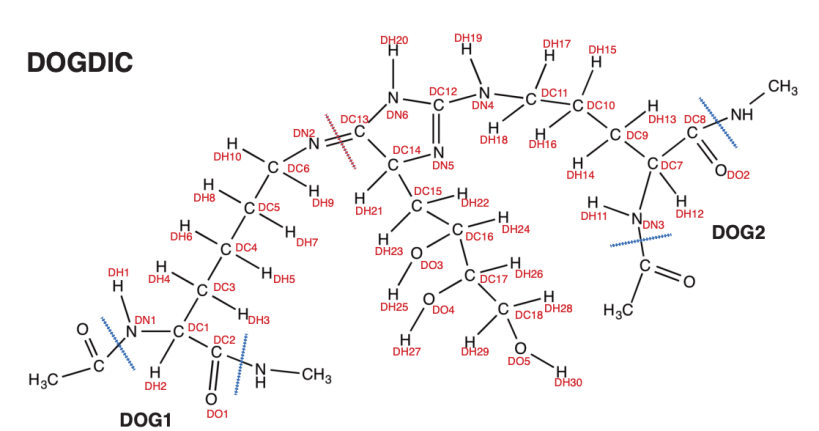
The following files must change:
atomtypes.atp
This file holds all atom types along with the atomic mass. Pretty simple, the type is used in the second column of an .itp file after pdb2gmx and in the second column in aminoacids.rtp for pdb2gmx production. Below is a short example of some atoms from the DOGDIC structure (above).
DH1 1.00800 DH2 1.00800 DH3 1.00800 ... DO1 16.00000 DO2 16.00000 DO3 16.00000 ... DN1 14.01000 DN2 14.01000 DN3 14.01000 .... DC1 12.01000 DC2 12.01000 DC3 12.01000 ...
aminoacids.r2b
Both parts of the crosslink (an arginine derived and lysine derived) will be centre residues. If you want PTM amino acids as part of the N- and C- terminus, additional entries to this file and further residue entries to aminoacids.rtp must be made. For example, valine has a central and N- and C- entries, whereas my PTM molecules will always have residues on a backbone either side.
; main N-ter C-ter 2-ter VAL VAL NVAL CVAL - MOD1 MOD1 - - - MOD2 MOD2 - - - GOD1 GOD1 - - - GOD2 GOD2 - - - DOG1 DOG1 - - - DOG2 DOG2 - - - GLP1 GLP1 - - - GLP2 GLP2 - - -
aminoacids.rtp
There are alternative means of implementing a new residue type without changing this file. However, I’ve always kept a backup of the original and worked on adding post-modified side chains into the backend force field files. Do this with caution.
Each [ atoms ] row requires four columns: atom name, atom type (as found in the atomtypes.atp file), atom charge, and atom charge group. Atom type matches all forcefield atom data e.g., bonded (ffbonded.itp), nonbonded (ffnonbonded.itp) and atomic weight (atomtypes.atp). Be warned, the quick and dirty approach of using matching atom name and atom type e.g., DO1 DO1 -0.416 5 comes with a few painful problems a lot later; without the significant hassle of converting the .gro file back into a pdb file and adding CONECT data (or by supplying a PSF file with the PDF), the likes of VMD will not understand how to render the bond connectivity. However, it is also worth noting that GROMACS pdb2gmx will fail if an atom name is used more than once in a residue. Atom types can be identical, atom names cannot.
So, how do we name atoms whilst maintaining the correct atom bond rendering in VMD? It turns out, after a little trial and error, all you need to do is begin each atom name with the atomic element symbol. For example, CD is the atom name for the CT atom type, however, you could also replace it with C99 or any label beginning with a C. Please note: this was from trial and error and may work in only a few cases and I’m yet to find official VMD documentation to back this up.
The official breakdown of the aminoacids.rtp file can be found here and lots here, both are well worth the read and go beyond the scope of this simple tutorial.
As I’m dealing with a PTM crosslinked protein, I’m adding two new side chains to the aminoacids.rtp file. The first part of the crosslink:
;DOGDIC crosslink [ DOG1 ] [ atoms ] N DN1 -0.3394 1 HN DH1 0.2125 2 CD1 DC1 -0.0601 3 HD1 DH2 0.0916 4 C DC2 0.5414 5 OD1 DO1 -0.4646 6 CD3 DC3 -0.0126 7 HD31 DH3 0.0216 8 HD32 DH4 0.0216 9 CD4 DC4 0.0114 1 HD41 DH5 0.0034 11 HD42 DH6 0.0034 12 CD5 DC5 -0.0006 13 HD51 DH7 0.0190 14 HD52 DH8 0.0190 15 CD6 DC6 0.1004 16 HD61 DH9 0.0323 17 HD62 DH10 0.0323 18 ND2 DN2 -0.6124 19 [ bonds ] N HN N CD1 CD1 HD1 CD1 C C OD1 CD1 CD3 CD3 HD31 CD3 HD32 CD3 CD4 CD4 HD41 CD4 HD42 CD4 CD5 CD5 HD51 CD5 HD52 CD5 CD6 CD6 HD61 CD6 HD62 CD6 ND2 -C N [ impropers ]
The second part of the crosslink:
[ DOG2 ] [ atoms ] N DN3 -0.4625 1 HN DH11 0.2490 2 CD7 DC7 -0.0118 3 HD1 DH12 0.0889 4 C DC8 0.6382 5 OD2 DO2 -0.5468 6 CD9 DC9 -0.0632 7 HD21 DH13 0.0303 8 HD22 DH14 0.0303 9 CD10 DC10 0.0127 10 HD31 DH15 0.0329 11 HD32 DH16 0.0329 12 CD11 DC11 0.0306 13 HD41 DH17 0.0837 14 HD42 DH18 0.0837 15 ND4 DN4 -0.7517 16 HD5 DH19 0.3830 17 CD12 DC12 0.7837 18 ND5 DN5 -0.6837 19 ND6 DN6 -0.6853 20 HD6 DH20 0.3616 21 CD13 DC13 0.5005 22 CD14 DC14 0.2385 23 HD7 DH21 0.0570 24 CD15 DC15 -0.0161 25 HD81 DH22 0.0576 26 HD82 DH23 0.0576 27 CD16 DC16 0.0435 28 HD9 DH24 0.0764 29 OD3 DO3 -0.7057 30 HD10 DH25 0.4552 31 CD17 DC17 0.1044 32 HD11 DH26 0.0965 33 OD4 DO4 -0.5962 34 HD12 DH27 0.4162 35 CD18 DC18 0.1135 36 H1 DH28 0.0353 37 H2 DH29 0.0353 38 OD5 DO5 -0.5938 39 HD13 DH30 0.3676 40 [ bonds ] N HN N CD7 CD7 HD1 CD7 C C OD2 CD7 CD9 CD9 HD21 CD9 HD22 CD9 CD10 CD10 HD31 CD10 HD32 CD10 CD11 CD11 HD41 CD11 HD42 CD11 ND4 ND4 HD5 ND4 CD12 CD12 ND5 CD12 ND6 ND6 HD6 ND5 CD14 ND6 CD13 CD13 CD14 CD14 HD7 CD14 CD15 CD15 HD81 CD15 HD82 CD15 CD16 CD16 HD9 CD16 OD3 OD3 HD10 CD16 CD17 CD17 HD11 CD17 OD4 OD4 HD12 CD17 CD18 CD18 HD1 CD18 HD2 CD18 OD5 OD5 HD13 -C N [ impropers ]
The –C indicates that an atom labelled C in the preceding residue will form a bond with N from this residue. As such, you’ll also need a C in this residue for the next residue along to form a peptide bond. Alternatively, you can remove this entry and build the bond by hand in the corresponding .itp after running gmx pdb2gmx. However, pdb2gmx is likely to view this residue as standalone peptide and therefore request terminal residue information. Providing as much information pre-pdb2gmx helps mitigate potential errors later.
specbond.dat
Bonds that cross between polypeptides and don’t make up the backbone, for example, a cystine bond are defined in specbond.dat. The file is in the directory that contains all forcefield directories and thus the file’s content is shared amongst each. For example, a cystine bond:
CYS SG 1 CYS SG 1 0.2 CYS2 CYS2
And the bond information for my PTM crosslinks:
MOD1 NM2 1 MOD2 CM6 1 0.125 MOD1 MOD2 GOD1 NG1 1 GOD2 CG12 1 0.125 GOD1 GOD2 DOG1 ND2 1 DOG2 CD13 1 0.125 DOG1 DOG2
Make sure you use the residue atom name (not the atom type). The distance (0.125) is the bond equilibrium distance and is taken from the QM calculation.
residuetypes.dat
This file ensures that GROMACS gmx commands recognise your new residue as part of a protein (or RNA or DNA). Using the residue name, I’ve added the following entries:
DOG1 Protein DOG2 Protein GOD1 Protein GOD2 Protein MOD1 Protein MOD2 Protein
aminoacids.hdb
This file is a pain to build but it will ensure the correct hydrogen bond behaviour when using pdb2gmx. Let’s take a look at lysine as a simple example. The number 7 next to the residue name LYS indicates 7 heavy atoms that require hydrogens to bond with. The first column (red) is the number of hydrogen bonds assigned to the first denoted heavy atom in each entry (green, that runs i, j, k and l, and depends on the bond geometry). The third column denotes atom names (purple). When more than one hydrogen atom is required (e.g., the third to fifth row) a number is added to the hydrogen name in the third column e.g., HB1, HB2, HG1, HG2… HZ1, HZ2, HZ3.
LYS 7 1 1 H N -C CA 1 5 HA CA N CB C 2 6 HB CB CA CG 2 6 HG CG CB CD 2 6 HD CD CG CE 2 6 HE CE CD NZ 3 4 HZ NZ CE CD
The second column is bond-type and this is where it gets a little painful:
1 – One planar hydrogen e.g., rings or peptide bonds. Spot the first line: N -C CA, hydrogen is attributed to N as you would expect.
2 – One single hydrogen e.g., hydroxyl group.
3 – Two planar hydrogens e.g., -C=CH2 or amides -C(=O)NH2.
4 – Two or three tetrahedral hydrogens e.g., -CH3. Down the side chain of lysine CD-CE-NZ are three hydrogens forming a tetrahedral with the NZ atom.
5 – One tetrahedral hydrogen e.g., C3CH. In all amino acids, the arrangement N-CA(-CB)-C forms a tetrahedral off the CA atom.
6 – Two tetrahedral hydrogens e.g., C-CH2-C. In lysine, CB, CG, CD and CE all have two type-6 hydrogens. This runs down the length of side chains in the likes of arginine, lysine, histidine etc.
(type 7, 10 and 11 are for water molecules).
The first cross-link residue:
DOG1 6 1 5 HD1 CD1 C N CD3 1 1 HN N -C CD1 2 6 HD3 CD3 CD1 CD4 2 6 HD4 CD4 CD3 CD5 2 6 HD5 CD5 CD4 CD6 2 6 HD6 CD6 CD5 ND2
ffbonded.itp
I won’t discuss the bonded parameters in too much detail, it’s beyond the scope of this tutorial. Prior to considering MD simulations, one would have had to acquire such parameters from either the literature, experimentation, a QM step, or a series of structural optimisation steps. In my example, I am adding to the AMBER forcefield family, therefore I’ve derived my bonded parameters by calculating the Hessian of the electronic structure with ForceGen (software I wrote a few years ago). Alternatively, one could try using ANTECHAMBER, but I have little experience using it.
Whichever method you use the distance and angle force constants are stored in ffbonded.itp. Atoms are denoted using their type (not name). Columns i and j denote the atom bond connectivity, the third column is the equilibrium bond distance in nm and the fifth column is the force constant in kJ/mol.
[ bondtypes ] ; i j func b0 kb ;DOGDIC DN1 DH1 1 0.099 171548.261 DN1 DC1 1 0.144 121887.165 DC1 DH2 1 0.108 153399.965 DC1 DC2 1 0.153 89908.146 DC2 DO1 1 0.122 315482.806 DC1 DC3 1 0.154 84206.191 DC3 DH3 1 0.108 45856.944 DC3 DH4 1 0.109 44286.368 DC3 DC4 1 0.153 93448.692 DC4 DH5 1 0.108 152748.149 DC4 DH6 1 0.109 132493.413 DC4 DC5 1 0.153 95415.177 DC5 DH7 1 0.109 89672.313 DC5 DH8 1 0.109 178049.914 DC5 DC6 1 0.154 105352.500 DC6 DH9 1 0.108 110735.315 DC6 DH10 1 0.109 124359.421 DC6 DN2 1 0.144 63531.021 N DC2 1 0.133 176436.798 C DN1 1 0.135 162246.033 DN2 DC13 1 0.125 186307.095 N DC8 1 0.133 176423.750 DC8 DO2 1 0.121 325547.562 DC8 DC7 1 0.153 96705.913 DC7 DH12 1 0.108 115294.316 DC7 DN3 1 0.146 116086.542 DN3 DH11 1 0.100 139423.472 DN3 C 1 0.134 190916.125 DC7 DC9 1 0.154 84120.874 DC9 DH13 1 0.108 115947.250 DC9 DH14 1 0.109 127095.404 DC9 DC10 1 0.153 105841.202 DC10 DH15 1 0.109 405220.497 DC10 DH16 1 0.108 138813.491 DC10 DC11 1 0.153 109300.079 DC11 DH17 1 0.108 107670.503 DC11 DH18 1 0.108 67798.216 DC11 DN4 1 0.146 121228.221 DN4 DH19 1 0.100 221292.284 DN4 DC12 1 0.137 160604.034 DC12 DN5 1 0.126 266534.962 DC12 DN6 1 0.138 153142.711 DN6 DH20 1 0.100 82736.086 DN6 DC13 1 0.140 99438.630 DC13 DC14 1 0.155 95552.445 DC14 DN5 1 0.146 101817.935 DC14 DH21 1 0.109 138339.658 DC14 DC15 1 0.153 53330.327 DC15 DH22 1 0.108 149115.074 DC15 DH23 1 0.108 150363.975 DC15 DC16 1 0.153 106887.426 DC16 DH24 1 0.108 85574.655 DC16 DO3 1 0.142 140524.412 DO3 DH25 1 0.095 238922.320 DC16 DC17 1 0.153 96996.331 DC17 DH26 1 0.109 123848.916 DC17 DO4 1 0.140 87739.129 DO4 DH27 1 0.095 102129.168 DC17 DC18 1 0.152 110262.387 DC18 DH28 1 0.109 74839.712 DC18 DH29 1 0.109 136564.454 DC18 DO5 1 0.139 69438.079 DO5 DH30 1 0.095 96059.354
There are too many bond angle force constants to display them all, however, here is a reduced example:
[ angletypes ] ; i j k func th0 cth ;DOGDIC C DN3 DC7 1 126.671 292.174 DH23 DC15 DC16 1 107.002 203.476 DC15 DC16 DH24 1 109.717 270.997 DC11 DN4 DH19 1 114.348 107.553 DH20 DN6 DC13 1 119.864 254.587 DH22 DC15 DC16 1 110.645 96.376 DH23 DC15 DC16 1 107.002 203.476 DH13 DC9 DC10 1 111.124 193.321 DH14 DC9 DC10 1 109.541 121.168 CT N DC2 1 124.326 278.510 H N DC2 1 117.820 262.167 N DC2 DC1 1 115.327 361.208 N DC2 DO1 1 123.562 356.408 DO1 DC2 DC1 1 121.111 347.062 DC2 DC1 DH2 1 108.461 113.542 DC2 DC1 DC3 1 112.632 263.616
Columns i, j and k denote the atom bond connectivity with the angle centred on j, the fifth column is the equilibrium bond angle in degrees and the sixth column is the force constant in kJ/mol.
ffnonbonded.itp
As with the bonded force constants, the derivation of non-bonded forces is beyond the scope of this tutorial. I took values from atoms with very similar structural chemistry to those already parameterised in the AMBER forcefield. However, if your structure is a bit beyond a modified amino acid, parameterisation using e.g, QM energy surface scans may be required. The DOGDIC non-bonded values are:
[ atomtypes ] ; name at.num mass charge ptype sigma epsilon DN1 7 14.01 0.0000 A 3.25000e-01 7.11280e-01 ;* DN2 7 14.01 0.0000 A 3.25000e-01 7.11280e-01 ;* DN3 7 14.01 0.0000 A 3.25000e-01 7.11280e-01 ;* DN4 7 14.01 0.0000 A 3.25000e-01 7.11280e-01 ;* DN5 7 14.01 0.0000 A 3.25000e-01 7.11280e-01 ;* DN6 7 14.01 0.0000 A 3.25000e-01 7.11280e-01 ;* DO1 8 16.00 0.0000 A 2.95992e-01 8.78640e-01 ;* DO2 8 16.00 0.0000 A 2.95992e-01 8.78640e-01 ;* DO3 8 16.00 0.0000 A 3.06647e-01 8.80314e-01 ;* DO4 8 16.00 0.0000 A 3.06647e-01 8.80314e-01 ;* DO5 8 16.00 0.0000 A 3.06647e-01 8.80314e-01 ;* DC1 6 12.01 0.0000 A 3.39967e-01 4.57730e-01 ;* DC2 6 12.01 0.0000 A 3.39967e-01 3.59824e-01 ;* DC3 6 12.01 0.0000 A 3.39967e-01 4.57730e-01 ;* DC4 6 12.01 0.0000 A 3.39967e-01 4.57730e-01 ;* DC5 6 12.01 0.0000 A 3.39967e-01 4.57730e-01 ;* DC6 6 12.01 0.0000 A 3.39967e-01 4.57730e-01 ;* DC7 6 12.01 0.0000 A 3.39967e-01 4.57730e-01 ;* DC8 6 12.01 0.0000 A 3.39967e-01 3.59824e-01 ;* DC9 6 12.01 0.0000 A 3.39967e-01 4.57730e-01 ;* DC10 6 12.01 0.0000 A 3.39967e-01 4.57730e-01 ;* DC11 6 12.01 0.0000 A 3.39967e-01 4.57730e-01 ;* DC12 6 12.01 0.0000 A 3.39967e-01 3.59824e-01 ;* DC13 6 12.01 0.0000 A 3.39967e-01 3.59824e-01 ;* DC14 6 12.01 0.0000 A 3.39967e-01 4.57730e-01 ;* DC15 6 12.01 0.0000 A 3.39967e-01 4.57730e-01 ;* DC16 6 12.01 0.0000 A 3.39967e-01 4.57730e-01 ;* DC17 6 12.01 0.0000 A 3.39967e-01 4.57730e-01 ;* DC18 6 12.01 0.0000 A 3.39967e-01 4.57730e-01 ;* DH1 1 1.008 0.0000 A 1.06908e-01 6.56888e-02 ;* DH2 1 1.008 0.0000 A 2.47135e-01 6.56888e-02 ;* DH3 1 1.008 0.0000 A 2.64953e-01 6.56888e-02 ;* DH4 1 1.008 0.0000 A 2.64953e-01 6.56888e-02 ;* DH5 1 1.008 0.0000 A 2.64953e-01 6.56888e-02 ;* DH6 1 1.008 0.0000 A 2.64953e-01 6.56888e-02 ;* DH7 1 1.008 0.0000 A 2.64953e-01 6.56888e-02 ;* DH8 1 1.008 0.0000 A 2.64953e-01 6.56888e-02 ;* DH9 1 1.008 0.0000 A 1.95998e-01 6.56888e-02 ;* DH10 1 1.008 0.0000 A 1.95998e-01 6.56888e-02 ;* DH11 1 1.008 0.0000 A 1.06908e-01 6.56888e-02 ;* DH12 1 1.008 0.0000 A 2.47135e-01 6.56888e-02 ;* DH13 1 1.008 0.0000 A 2.64953e-01 6.56888e-02 ;* DH14 1 1.008 0.0000 A 2.64953e-01 6.56888e-02 ;* DH15 1 1.008 0.0000 A 2.64953e-01 6.56888e-02 ;* DH16 1 1.008 0.0000 A 2.64953e-01 6.56888e-02 ;* DH17 1 1.008 0.0000 A 2.47135e-01 6.56888e-02 ;* DH18 1 1.008 0.0000 A 2.47135e-01 6.56888e-02 ;* DH19 1 1.008 0.0000 A 1.06908e-01 6.56888e-02 ;* DH20 1 1.008 0.0000 A 1.06908e-01 6.56888e-02 ;* DH21 1 1.008 0.0000 A 2.47135e-01 6.56888e-02 ;* DH22 1 1.008 0.0000 A 2.47135e-01 6.56888e-02 ;* DH23 1 1.008 0.0000 A 2.47135e-01 6.56888e-02 ;* DH24 1 1.008 0.0000 A 2.47135e-01 6.56888e-02 ;* DH25 1 1.008 0.0000 A 0.00000e+00 0.00000e+00 ;* DH26 1 1.008 0.0000 A 2.47135e-01 6.56888e-02 ;* DH27 1 1.008 0.0000 A 0.00000e+00 0.00000e+00 ;* DH28 1 1.008 0.0000 A 2.47135e-01 6.56888e-02 ;* DH29 1 1.008 0.0000 A 2.47135e-01 6.56888e-02 ;* DH30 1 1.008 0.0000 A 0.00000e+00 0.00000e+00 ;*
Further information on modifying forcefield files can be found at the GROMACS home page.
Building the initial geometry
I’ll be adding the crosslink to the collagen protein structure using Avogadro. I’ll then use pdb2gmx to convert the PDF file to a GRO file whilst generating the topology data.
There are a few points to note when modifying structures in Avogadro. Swapping a group of existing atoms for a different group e.g., hydroxyl for carbonyl, adds atom line entries within the position of the residue in the file. The replaced atoms will usually (there are probably exceptions that I’m unaware of) be denoted with HETATM. Adding a new residue adds atoms lines to the bottom of the file. If the residue is recognised e.g., a standard amino acid, then ATOM is used, otherwise, HETATM is used.
HETATM means hetero atom and is applied to non-standard residues of protein, DNA or RNA, as well as atoms in other kinds of groups, such as carbohydrates, ligands, solvents, and metals.
Firstly, I took a geometry optimised structure from an earlier simulation of human collagen as my wildtype. A collagen protein is made up of three polypeptide chains. This particular system has nine proteins i.e., 27 polypeptide chains. We’ll be crosslinking two polypeptide chains in one protein.

Ignore how the tightly bound collagen peptides have detached from the main fibral, what you’re seeing is a periodic boundary artefact. The file is in a .gro format and the atom layout is compatible with my AMBER forcefield of choice. To go from a .gro to .pdb I simply use editconf:
gmx editconf -f initial.gro -o initial.pdb
To sanity check that I won’t have any problems with the elements of this structure that won’t be modified I run it through pdb2gmx:
gmx pdb2gmx -f initial.pdb -o test.gro
I select the AMBER forcefield of choice and then I select no water. If this back-and-forth approach works, then you have an overall structure that won’t be causing any problems. It’s worth keeping an eye on the coordinate values. Moving between .gro and .pdb using gmx should preserve the coordinate values:
As a .pdb (stored in angstroms):
ATOM 1 N PRO 1 0.170 20.430 13.400 1.00 0.00
As a .gro (stored in nanometres):
1PRO N 1 0.017 2.043 1.340 0.0040 0.1964 -0.0452
It is important to note that Avogadro preserves the coordinates when saving as a .pdf:
ATOM 1 N PRO 1 0.170 20.430 13.400 1.00 0.00 N
This is important to remember as we’ll be removing a single protein (three polypeptide chains), modifying it, then adding it back into the overall structure.
Next, I remove the first three polypeptide chains that make up a complete protein. If I’ve taken this directly from the .gro file I’ll need to pass it through gmx editconf to get a .pdb file. When loaded into Avogadro it looks like:
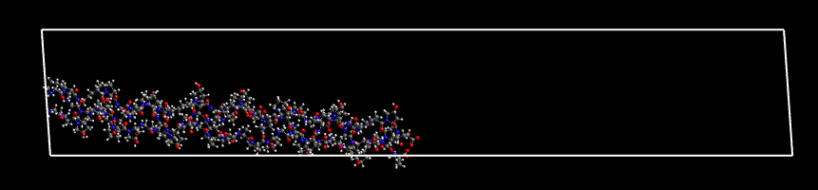
Given the box dimensions, you’ll now begin to appreciate why the final .gro coordinate file looked as though the tightly packed protein had come apart.
Begin by identifying which part of the collagen protein will be modified along with the corresponding complete residue (including the N-C(H)-C(=O) backbone). Be very careful that you remove the correct backbone atoms. Check using Atom Properties in Avogadro and the atom index in the .pdb file or rather than using Avogadro to remove atoms, simply open up the .pdb file and delete both identified residues.
Having removed the complete amino acid, I’ve highlighted the neighbouring residue backbone C Beta and N atoms.

The empty space left behind is very noticeable when a complete residue has been removed from both neighbouring polypeptide chain (a crosslink requires two placements along the backbone).

It’s a good idea to save the file at this point. I’ll be adding different crosslinks for further simulations at a later date.
Adding a crosslink
With the collagen prepared the next steps are creating the crosslink and adding the crosslink into the collagen framework.
Creating the crosslink is fairly simple using Avogadro’s set of Draw Tools. It’s best to do this in a separate Avogadro window so one can then perform a basic energy minimisation on just the crosslink. When the structure is prepared (save and store) copy the crosslink into the collagen framework and orientate it near the peptide backbone gap.

Before connecting the crosslink to the collagen backbone it is very important to fix the coordinates of the collagen so they don’t adjust when we perform a simple energy minimisation in Avogadro. Using your mouse highlight all collagen atoms to the left of the gap whilst leaving a few of the immediate backbone atoms unselected. Selection Extensions->Molecular Mechanics->Fix Selected Atoms. Repeat this for all atoms to the right of the gap. Draw bonds from the crosslink to the collagen peptide. Do not perform this the other way around as we want to preserve the labelling of the neighbouring peptide backbone. Also, ensure that Adjust Hydrogens is turned OFF.

With the long bonds in place, it’s time to position the crosslink in place by performing an energy minimisation within Avogadro. This may take ten to fifteen minutes depending on your hardware and on how close you positioned the crosslink to the peptide backbone.

With the crosslink attached to neighbouring peptide backbones and inserted in place using energy minimisation steps, it’s time to manipulate the atom labelling so the file is recognised by gmx pdb2gmx. Save the new collagen structure as a pdb file.
Load the pdb file into VMD and position the content of the pdb file in a text editor (VI in this instance) and the VMD console output and the VMD display next to one another. Using the pick command we can use the mouse to left-click select on each atom. The index value plus one (VMD counts atoms from 0, much like C/C++) is the location of the atom in the pdb file.
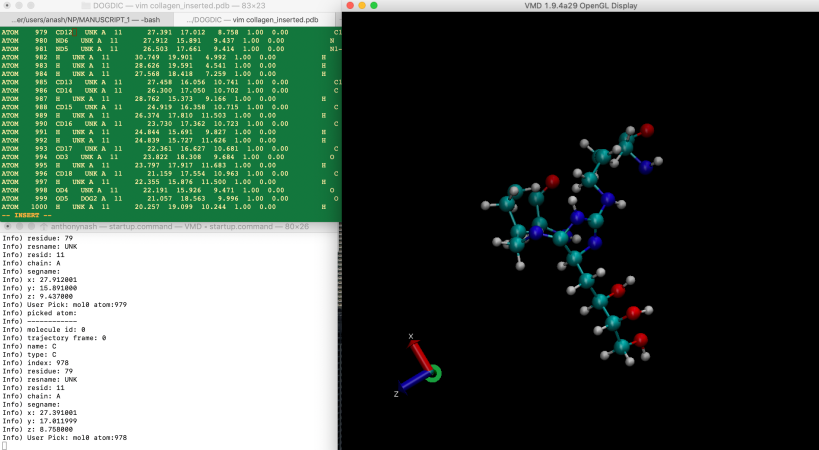
Go through each heavy atom (ignore the hydrogens), identify their position in the file using their index and rename the atom name according to the aminoacids.rtp entry. Don’t forget to rename the residue name from UNK to either DOG1 or DOG2. Avogadro will have placed both parts of the crosslink (as we use a special bond in specbond.dat to connect the two residues) together. After completing all atom names and residue names, the output will look similar to:
ATOM 946 ND1 DOG1 89 27.956 21.534 10.750 1.00 0.00 N ATOM 947 CD1 DOG1 89 29.129 21.166 11.568 1.00 0.00 C1- ATOM 948 CD2 DOG1 89 30.431 21.826 11.062 1.00 0.00 C ATOM 949 OD1 DOG1 89 30.740 21.681 9.890 1.00 0.00 O ATOM 950 CD3 DOG1 89 31.056 19.409 11.689 1.00 0.00 C1- ATOM 951 CD4 DOG1 89 30.618 18.058 12.310 1.00 0.00 C ATOM 952 CD5 DOG1 89 29.074 18.071 12.547 1.00 0.00 C ATOM 953 CD6 DOG1 89 28.417 16.706 12.884 1.00 0.00 C ATOM 954 ND2 DOG1 89 28.351 15.920 11.674 1.00 0.00 N1- ATOM 955 H DOG1 89 28.927 21.473 12.612 1.00 0.00 H ATOM 956 H DOG1 89 30.691 19.193 10.656 1.00 0.00 H ATOM 957 H DOG1 89 30.474 19.997 12.242 1.00 0.00 H ATOM 958 H DOG1 89 31.107 17.885 13.292 1.00 0.00 H ATOM 959 H DOG1 89 30.928 17.254 11.608 1.00 0.00 H ATOM 960 H DOG1 89 28.563 18.488 11.653 1.00 0.00 H ATOM 961 H DOG1 89 28.886 18.771 13.389 1.00 0.00 H ATOM 962 H DOG1 89 27.393 16.840 13.291 1.00 0.00 H ATOM 963 H DOG1 89 29.023 16.183 13.653 1.00 0.00 H ATOM 964 H DOG1 89 27.465 20.755 10.280 1.00 0.00 H ATOM 965 ND3 DOG2 90 28.735 20.268 5.308 1.00 0.00 N ATOM 966 CD7 DOG2 90 30.164 20.410 5.701 1.00 0.00 C ATOM 967 CD8 DOG2 90 30.633 21.872 5.503 1.00 0.00 C ATOM 968 OD2 DOG2 90 30.547 22.348 4.379 1.00 0.00 O ATOM 969 CD9 DOG2 90 30.405 19.949 7.040 1.00 0.00 C ATOM 970 H DOG2 90 29.675 20.166 7.842 1.00 0.00 H ATOM 971 H DOG2 90 31.411 19.928 7.379 1.00 0.00 H ATOM 972 CD10 DOG2 90 30.178 18.482 6.818 1.00 0.00 C ATOM 973 H DOG2 90 30.066 17.763 5.977 1.00 0.00 H ATOM 974 H DOG2 90 29.688 19.441 6.544 1.00 0.00 H ATOM 975 CD11 DOG2 90 29.468 17.898 8.003 1.00 0.00 C ATOM 976 H DOG2 90 29.516 18.632 8.837 1.00 0.00 H ATOM 977 H DOG2 90 29.992 16.970 8.310 1.00 0.00 H ATOM 978 ND4 DOG2 90 28.076 17.632 7.696 1.00 0.00 N ATOM 979 CD12 DOG2 90 27.391 17.012 8.758 1.00 0.00 C1- ATOM 980 ND6 DOG2 90 27.912 15.891 9.437 1.00 0.00 N ATOM 981 ND5 DOG2 90 26.503 17.661 9.414 1.00 0.00 N1- ATOM 982 H DOG2 90 30.749 19.901 4.992 1.00 0.00 H ATOM 983 H DOG2 90 28.626 19.591 4.541 1.00 0.00 H ATOM 984 H DOG2 90 27.568 18.418 7.259 1.00 0.00 H ATOM 985 CD13 DOG2 90 27.458 16.056 10.741 1.00 0.00 C1- ATOM 986 CD14 DOG2 90 26.300 17.050 10.702 1.00 0.00 C ATOM 987 H DOG2 90 28.762 15.373 9.166 1.00 0.00 H ATOM 988 CD15 DOG2 90 24.919 16.358 10.715 1.00 0.00 C ATOM 989 H DOG2 90 26.374 17.810 11.503 1.00 0.00 H ATOM 990 CD16 DOG2 90 23.730 17.362 10.723 1.00 0.00 C ATOM 991 H DOG2 90 24.844 15.691 9.827 1.00 0.00 H ATOM 992 H DOG2 90 24.839 15.727 11.626 1.00 0.00 H ATOM 993 CD17 DOG2 90 22.361 16.627 10.681 1.00 0.00 C ATOM 994 OD3 DOG2 90 23.822 18.308 9.684 1.00 0.00 O ATOM 995 H DOG2 90 23.797 17.917 11.683 1.00 0.00 H ATOM 996 CD18 DOG2 90 21.159 17.554 10.963 1.00 0.00 C ATOM 997 H DOG2 90 22.355 15.876 11.500 1.00 0.00 H ATOM 998 OD4 DOG2 90 22.191 15.926 9.471 1.00 0.00 O ATOM 999 OD5 DOG2 90 21.057 18.563 9.996 1.00 0.00 O ATOM 1000 H DOG2 90 20.257 19.099 10.244 1.00 0.00 H ATOM 1001 H DOG2 90 21.286 18.012 11.969 1.00 0.00 H ATOM 1002 H DOG2 90 20.231 16.938 10.957 1.00 0.00 H ATOM 1003 H DOG2 90 21.976 16.581 8.756 1.00 0.00 H ATOM 1004 H DOG2 90 23.906 17.817 8.826 1.00 0.00 H
The order of the atoms in a residue does not matter. The pdb2gmx will map the input atoms to the atoms in the aminoacid.rtp database on a residue by residue basis. Having already deleted two residues to fit the crosslink in, I divided this list by DOG1 and DOG2 placing them into their respective locations; this was essential, otherwise, pdb2gmx would form the backbone peptide bond between the wrong residues.
Generating GROMACS coordinate and topology files
This is perhaps the easiest step providing no mistakes have been made. Execute:
gmx pdb2gmx -f collagen.pdb -o crosslinked.gro -ignh
It’s essential that you ignore hydrogens (using -ignh). We didn’t adjust their atom name using VMD, rather, we are relying on the hydrogen bond database entry for DOG1 and DOG2.
Providing no errors occurred (I had a few warnings of long bonds, namely from having centre amino acids at the peptide termini; simply renaming PRO to CPRO and GLY to NGLY fixed these) you should have a .gro file and a .top file.
A final note on dihedrals. Whilst we didn’t provide any force constant values for dihedrals in the force field, gmx pdb2gmx will have built dihedral bond descriptions in the topology file. Those must be removed from the topology file if you hope to see this work with gmx grompp. However, I recommend you populate the dihedral angles using the same steps for bond length and bond angle.
